In this section we will write a web application able to reverse, complement, or reverse-complement a DNA sequence. Starting from a DNA sequence, the reverse-complement operation enables to compute the sequence of the complementary strand, as already discussed in section 4-7 of this book where we have also provided a simple code able to achieve this operation. Building up on this code, in section 4-12 we have written a PHP function able to perform the task.
Single sequence version
We will now leverage on a slightly improved version of this function, which supports IUPAC characters, to build a web application that can compute the reverse, complement, or reverse-complement of sequences provided by users, in a web context.
We will also use the sequence breaker function seqbreak() to introduce a break every 80 nucleotides, to format the output sequence.
Using the previously written functions, stored in a functions.php file that is then imported in the script file with an “include” statement, will allow the script.php code to be extremely compact.
The application will support the FASTA format, the IUPAC code for degenerate sequences, and will have options to select the kind of transformation (reverse, complement or reverse-complement) to be applied on the input sequence.
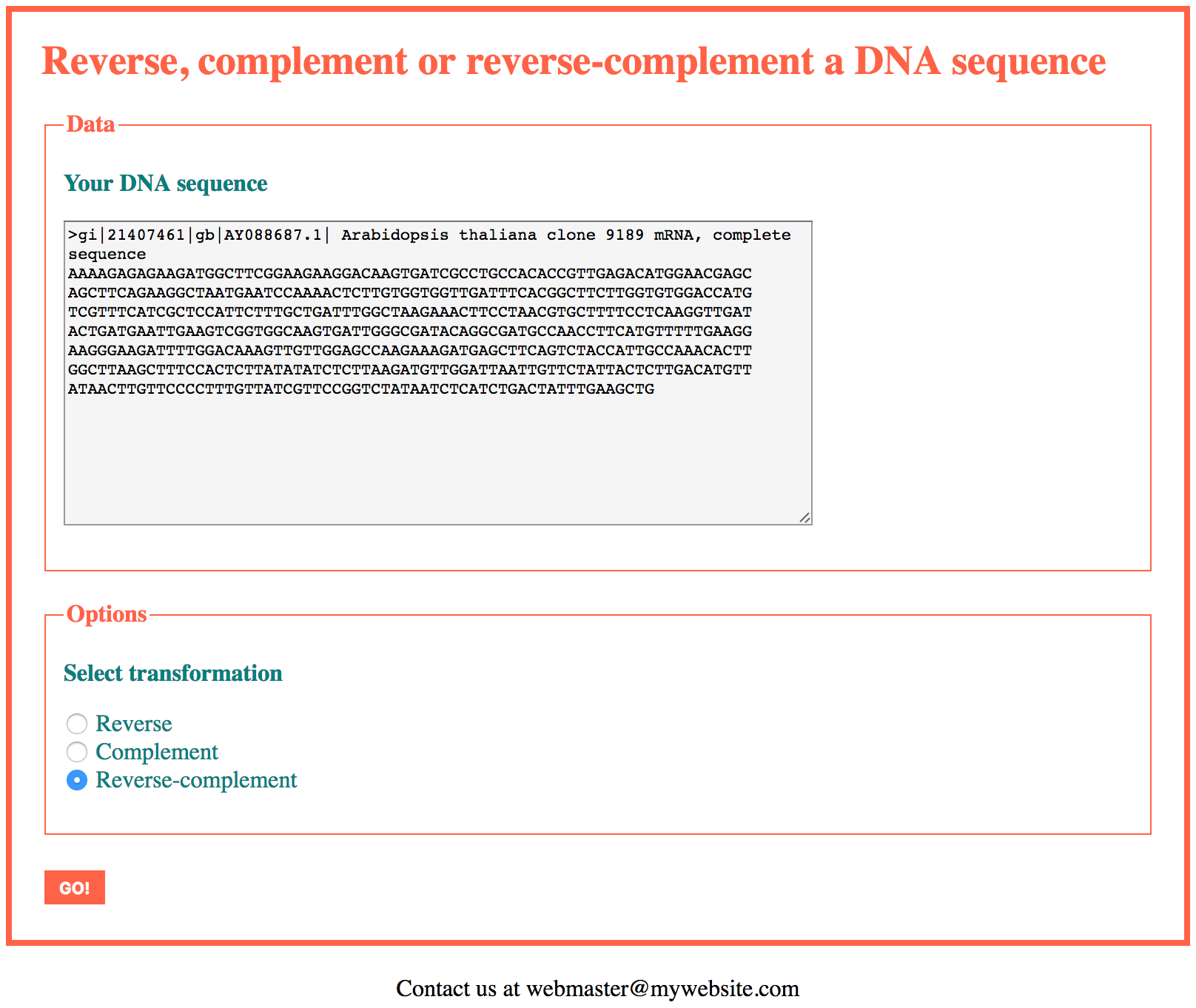
In this first version of the application we will accept as input a single DNA sequence. At the end of this section we propose a version able to handle several sequences.
The code
As usual, the code for the web application will be distributed across several files. The general structure is the same as the one of the application developed in the previous section. Directories names are in bold.
reverse-complement
index.php
script.php
html
header.html
footer.html
css
style.css
include
functions.php
header.html
|
1 2 3 4 5 6 7 8 9 10 11 12 |
<!DOCTYPE html> <html lang="en"> <head> <meta charset="UTF-8"> <title>A script to reverse-complement a DNA sequence</title> <link rel="stylesheet" href="css/style.css" type="text/css"> </head> <body> <div id="main-contents"> <h1>Reverse, complement or reverse-complement a DNA sequence</h1> |
footer.html
|
1 2 3 4 5 6 7 8 |
</div> <footer> Contact us at webmaster@mywebsite.com </footer> </body> </html> |
index.php
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 |
<?php echo file_get_contents("html/header.html"); ?> <form action="script.php" method="POST" enctype="multipart/form-data" id="revcomp_form"> <fieldset id="data"> <legend>Data</legend> <p> <label for="fasta_sequence">Your DNA sequence</label><br> </p> <p> <textarea name="fasta_sequence" id="fasta_sequence"></textarea> </p> </fieldset> <fieldset id="options"> <legend>Options</legend> <p> <span class="field-title" style="margin-bottom:10px;">Select transformation</span> </p> <p> <input type="radio" name="transformation" value="rev" id="rev"> <label for="rev" class="radio">Reverse</label><br> <input type="radio" name="transformation" value="comp" id="comp"> <label for="comp" class="radio">Complement</label><br> <input type="radio" name="transformation" value="revcomp" id="revcomp" checked> <label for="revcomp" class="radio">Reverse-complement</label> </p> </fieldset> <input type="submit" value="Go!"> </form> <?php echo file_get_contents("html/footer.html"); ?> |
functions.php
The reverse-complement function – revcomp() – used in this section is modified with respect to the one proposed in section 4-12 so as to support all IUPAC characters for nucleotides (A, C, G, T, U, R, Y, S, W, K, M, B, D, H, V, N, ., -). More specifically, the complement dictionary associative array was extended.
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 |
<?php function process_fasta($fasta_sequence, $mode="all"){ $fasta_lines = explode("\n", $fasta_sequence); $header = "> Generic"; // We will store the header line here during the next foreach cycle $sequence = ""; // We will store the sequence here during the next foreach cycle foreach($fasta_lines as $line){ // We strip possible whitespace (or other characters) from the beginning and end of the line $line = trim($line); if(preg_match("/^>/", $line)){ // If the line starts with a > it's the header line $header = $line; } elseif($line != ""){ $sequence = $sequence.$line; // We concatenate each new sequence line in the $sequence variable } } // At this point we should have the FASTA header in the $header variable // and the whole sequence in the $sequence variable // And now the return part, that depends on value of $mode if($mode == "all"){ return array($header, $sequence); } elseif($mode == "seq"){ return $sequence; } elseif($mode == "header"){ return $header; } else{ return "WARNING: process_fasta mode not supported"; } } function revcomp($sequence, $mode="revcomp"){ $complement_dict = array( "A" => "T", "T" => "A", "G" => "C", "C" => "G", "U" => "A", "R" => "Y", "Y" => "R", "S" => "W", "W" => "S", "K" => "M", "M" => "K", "B" => "D", "D" => "B", "H" => "V", "V" => "H", "N" => "N", "." => ".", "-" => "-" ); $nucleotides = str_split($sequence,1); // Let's compute the complement sequence first $complement_sequence = ""; foreach($nucleotides as $nucleotide){ $complement_sequence = $complement_sequence.$complement_dict[$nucleotide]; } // The complement sequence is now stored in the $complement_sequence variable $revcomp_sequence = strrev($complement_sequence); // This is the reverse complement sequence $reverse_sequence = strrev($sequence); // This is the reverse sequence // We return different things depending on the $mode (second optional argument of this function) // if we call the function with just one argument, the value of $mode will be the default, "revcomp" // additional supported values for the $mode argument are "comp" and "rev", see below // Note that when a function returns, it also exits, no more code inside the function is executed if($mode == "revcomp"){ return $revcomp_sequence; } elseif($mode == "comp"){ return $complement_sequence; } elseif($mode == "rev"){ return $reverse_sequence; } else{ // This part may help us in debugging code in which the function is used return "WARNING: revcomp mode not supported"; } } function seqbreak($sequence, $brlen=80, $brel="<br>\n"){ // $brel => breaking element $chars = str_split($sequence, 1); $i = 1; $out = ""; foreach($chars as $char){ if(is_int($i/$brlen)){ $out = $out.$char.$brel; } else{ $out = $out.$char; } $i++; } return $out; } ?> |
style.css
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 |
body{ width: 800px; margin-right:auto; margin-left:auto; } #main-contents{ border:4px solid tomato; margin-right:auto; margin-left:auto; margin-bottom: 20px; padding:20px; padding-top:0; } span.sequence{ font-family:courier; font-size:14px; } span.header{ font-size:14px; } label{ cursor:pointer; font-weight:bold; color:teal; } label.radio{ font-weight:normal; } span.field-title{ font-weight:bold; color:teal; } fieldset{ border:1px solid tomato; margin-bottom:20px; } legend{ font-weight:bold; color:tomato; } input[type=submit] { background-color: tomato; border: none; color: white; padding: 5px 10px; text-decoration: none; margin: 4px 2px; cursor: pointer; text-transform: uppercase; font-weight:bold; } h1{ color:tomato; font-size:1.7em; } textarea{ width:500px; height:200px; font-family:courier; background:whitesmoke; } input[type="text"]{ background:whitesmoke; } footer{ text-align:center; } |
script.php
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 |
<?php include("include/functions.php"); // We include the functions.php file $fasta_sequence = $_POST["fasta_sequence"]; // and grab the sequence submitted by the user through the web form // We call it fasta_sequence rather than sequence as it may well be a FASTA sequence. The name of the // field in the web form (index.php) was also changed accordingly $transformation = $_POST["transformation"]; // This will be revcomp (web form default), rev or comp $sequence = strtoupper(process_fasta($fasta_sequence, "seq")); // We use process_fasta() to extract the "raw" sequence // from the fasta_sequence and also make sure it is in uppercase characters $header = process_fasta($fasta_sequence, "header"); // We use process_fasta() to extract the header // We leverage on the revcomp() function to get the transformed sequence // Note how things are easy when we have a function to do the job $t_sequence = revcomp($sequence, $transformation); // We create the sequence ready for output by using the seqbreak() function // that introduces a break tag every 80 amino-acids $breaked_sequence = seqbreak($t_sequence); $out_sequence = "<span class=\"sequence\">".$breaked_sequence."</span>"; // We generate a $t_txt variable (transformation text) that is the complete version of the selected transformation // So we will be able to use "reverse-complement" instead of "revcomp", et-cetera. if($transformation == "revcomp"){ $t_txt = "reverse-complement"; } elseif($transformation == "rev"){ $t_txt = "reverse"; } elseif($transformation == "comp"){ $t_txt = "complement"; } // We create a new header for the transformed sequence by appending "reverse-complement", "reverse" or "complement" // to the original header, as appropriate $header_new = $header." - $t_txt"; // We now have all we need to provide an output to the user // We embed the output data within the same header and footer HTML code used in the web form // to ensure a consistent navigation experience and provide the feel that // everything takes place "in the same website" echo file_get_contents("html/header.html"); // Writing the header HTML to the output page echo "<h2>Transformed sequence</h2>\n"; echo "<p><strong>Selected transformation:</strong> $t_txt</p>"; echo "<p><span class=\"header\">$header_new</span><br>\n"; echo "$out_sequence</p>\n"; echo file_get_contents("html/footer.html"); // Writing the footer HTML to the output page ?> |
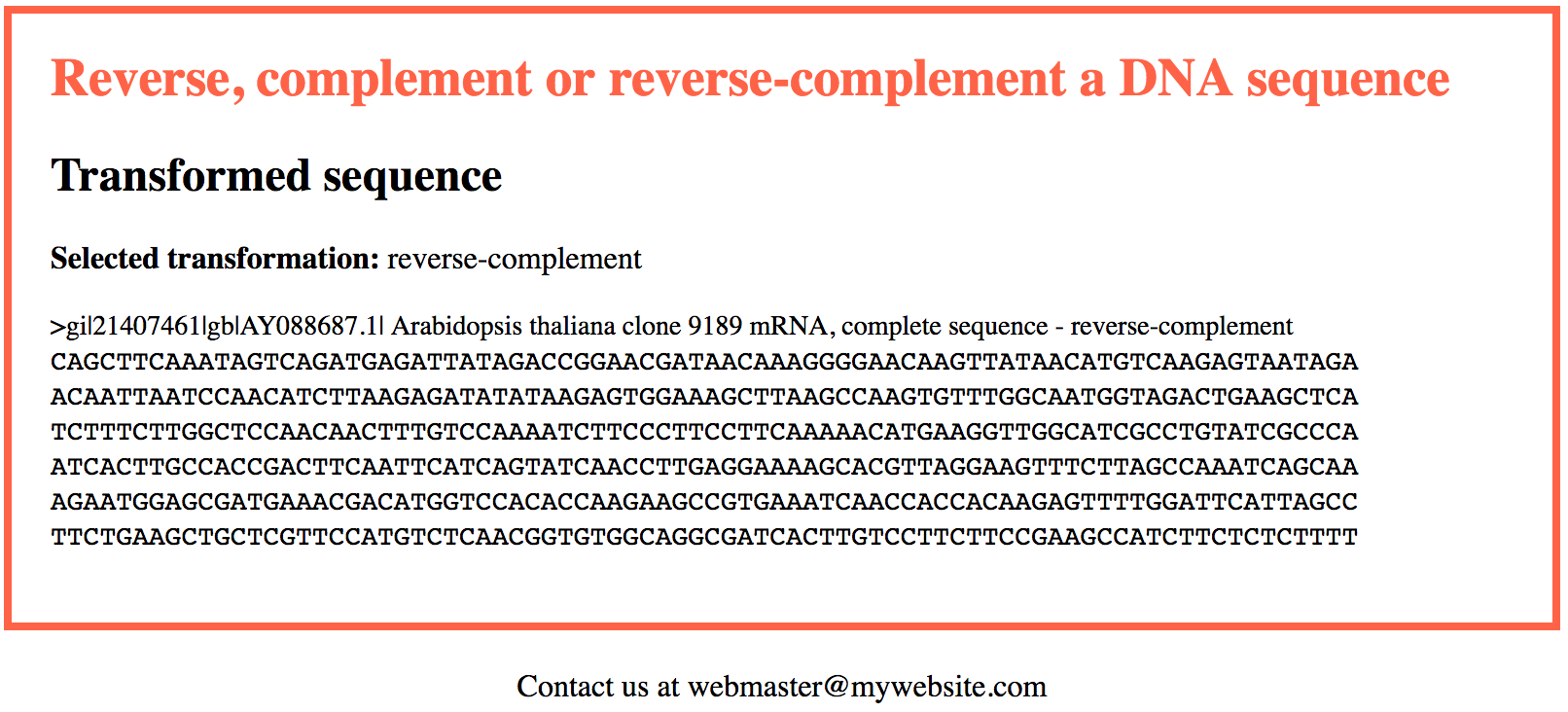
You may test the script live here.
Batch version
Let’s now write a version of this application able to process several FASTA sequences at the same time, in batch.
To accept multiple sequences in input, we will switch the FASTA processing function, from process_fasta() to fasta_sequences_to_array(). We have already written the code for both functions in section 4-12.
The header, footer and css files remain unchanged with respect to the single sequence version. In the web form (index.php), the only change will be the name of the text-area, namely “fasta_sequence” will be changed to “fasta_sequences”. The id of the text-area and the “for” attribute of the text-area label will also be adjusted to this new value.
index.php (batch version)
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 |
<?php echo file_get_contents("html/header.html"); ?> <form action="script.php" method="POST" enctype="multipart/form-data" id="revcomp_form"> <fieldset id="data"> <legend>Data</legend> <p> <label for="fasta_sequences">Your DNA sequence(s). Supports FASTA sequences in batch</label><br> </p> <p> <textarea name="fasta_sequences" id="fasta_sequences"></textarea> </p> </fieldset> <fieldset id="options"> <legend>Options</legend> <p> <span class="field-title" style="margin-bottom:10px;">Select transformation</span> </p> <p> <input type="radio" name="transformation" value="rev" id="rev"> <label for="rev" class="radio">Reverse</label><br> <input type="radio" name="transformation" value="comp" id="comp"> <label for="comp" class="radio">Complement</label><br> <input type="radio" name="transformation" value="revcomp" id="revcomp" checked> <label for="revcomp" class="radio">Reverse-complement</label> </p> </fieldset> <input type="submit" value="Go!"> </form> <?php echo file_get_contents("html/footer.html"); ?> |
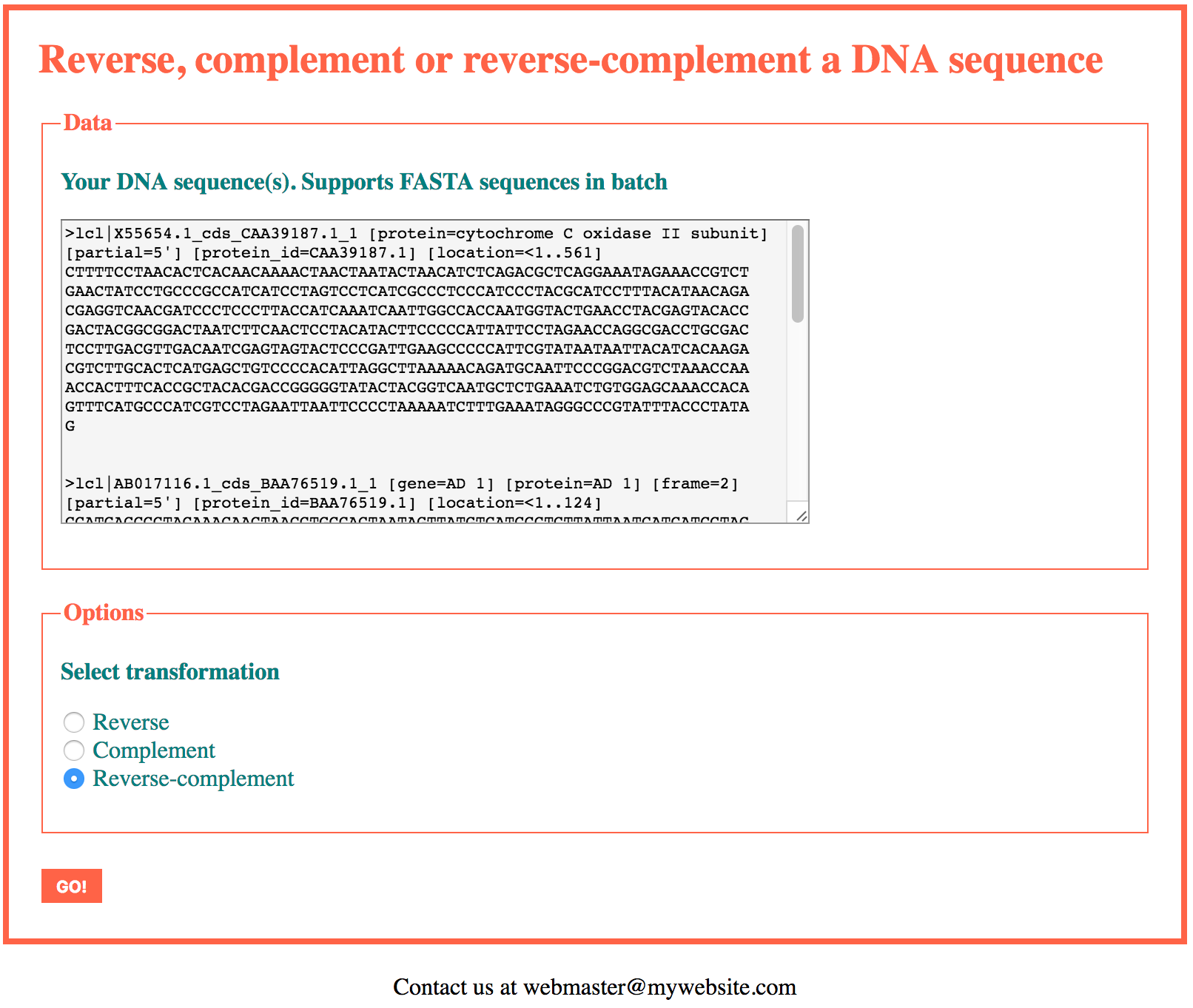
In the functions file we replace process_fasta() with fasta_sequences_to_array().
functions.php (batch version)
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 |
<?php function fasta_sequences_to_array($fasta_sequences){ // Takes a variable with FASTA sequences as input $lines = preg_split("/\n/", $fasta_sequences); // Individual lines to an array $seqs_array = array(); $sequence = ''; $header_line = ''; $i = 0; foreach($lines as $line){ if(preg_match("/^>/",$line)){ if($i != 0){ $seqs_array[] = array($header_line,$sequence); $sequence = ''; } $header_line = trim($line); $i++; } elseif($line != ''){ $sequence .= strtoupper(trim($line)); } } $seqs_array[] = array($header_line,$sequence); return $seqs_array; } function revcomp($sequence, $mode="revcomp"){ $complement_dict = array( "A" => "T", "T" => "A", "G" => "C", "C" => "G", "U" => "A", "R" => "Y", "Y" => "R", "S" => "W", "W" => "S", "K" => "M", "M" => "K", "B" => "D", "D" => "B", "H" => "V", "V" => "H", "N" => "N", "." => ".", "-" => "-" ); $nucleotides = str_split($sequence,1); // Let's compute the complement sequence first $complement_sequence = ""; foreach($nucleotides as $nucleotide){ $complement_sequence = $complement_sequence.$complement_dict[$nucleotide]; } // The complement sequence is now stored in the $complement_sequence variable $revcomp_sequence = strrev($complement_sequence); // This is the reverse complement sequence $reverse_sequence = strrev($sequence); // This is the reverse sequence // We return different things depending on the $mode (second optional argument of this function) // if we call the function with just one argument, the value of $mode will be the default, "revcomp" // additional supported values for the $mode argument are "comp" and "rev", see below // Note that when a function returns, it also exits, no more code inside the function is executed if($mode == "revcomp"){ return $revcomp_sequence; } elseif($mode == "comp"){ return $complement_sequence; } elseif($mode == "rev"){ return $reverse_sequence; } else{ // This part may help us in debugging code in which the function is used return "WARNING: revcomp mode not supported"; } } function seqbreak($sequence, $brlen=80, $brel="<br>\n"){ // $brel => breaking element $chars = str_split($sequence, 1); $i = 1; $out = ""; foreach($chars as $char){ if(is_int($i/$brlen)){ $out = $out.$char.$brel; } else{ $out = $out.$char; } $i++; } return $out; } ?> |
And here is the script.
A line of the code may deserve some explanation.
When we get the sequences from the web form, we convert them in an array ($seqs_array) with this structure:
[(seq1 header, seq1 sequence),(seq2 header, seq2 sequence),(seq3 header, seq3 sequence), etc…]
with the fasta_sequences_to_array() function.
In one line of code, we transfer this information to a second array, in which both the headers and the sequences are modified.
More specifically, we append to each header a text ($t_txt) with ” – reverse”, ” – complement”, or ” – reverse-complement”, depending on the transformation selected by the user.
The sequences themselves are also modified according to the selected transformation. HTML tags are also added to the sequence. In particular a break tag is added every 80 nucleotides with the seqbreak() function and the whole sequence is embedded within a span tag with a “sequence” class, which has a font-family:courier in the CSS file.
All of this is done in a single line of code within a foreach cycle:
|
1 2 3 4 5 |
foreach($seqs_array as $seq_array){ $seqs_array_t[] = array($seq_array[0]." - $t_txt", "<span class=\"sequence\">".seqbreak(revcomp(strtoupper($seq_array[1]), $transformation))."</span>"); } |
As it happens, to better understand the code, it should be read from right to left:
- The sequence ($seq_array[1]) is converted to uppercase, the only characters set the revcomp() function understands
- This uppercase sequence is passed as argument to revcomp() together with the selected transformation type ($transformation)
- The sequence transformed by revcomp() is added a break tag every 80 nucleotides with seqbreak()
- The sequence is then embedded within a span tag
- The header ($seq_array[0]) is added the appropriate text accounting for the transformation
- The transformed header and transformed and tagged sequence are the first and second element of an array
- This two elements array is added to the transformed sequences array $seqs_array_t
Read the comments in the code to better understand the flow.
script.php (batch version)
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 |
<?php include("include/functions.php"); // We include the functions.php file $fasta_sequences = $_POST["fasta_sequences"]; // and grab the sequences submitted by the user through the web form $transformation = $_POST["transformation"]; // This will be revcomp (web form default), rev or comp $seqs_array = fasta_sequences_to_array($fasta_sequences); $seqs_array_t = array(); // An empty array where the transformed sequences will be stored. t stays for "transformed" // Let's have some proper text values at hand to replace comp, revcomp and rev // in the user output if($transformation == "revcomp"){ $t_txt = "reverse-complement"; } elseif($transformation == "rev"){ $t_txt = "reverse"; } elseif($transformation == "comp"){ $t_txt = "complement"; } foreach($seqs_array as $seq_array){ // We transfer the sequences information from the original array to the transformed array. // Since we are at it: we turn each sequence in uppercase, do the transformation, apply a break tag every 80 nucleotides // and embed everything in a span tag with the class "sequence" so that the sequences will be in courier monoscpace font // note how all of this is done sequentially in a single line of code $seqs_array_t[] = array($seq_array[0]." - $t_txt", "<span class=\"sequence\">".seqbreak(revcomp(strtoupper($seq_array[1]), $transformation))."</span>"); } echo file_get_contents("html/header.html"); // Writing the header HTML to the output page echo "<h2>Transformed sequence(s)</h2>\n"; echo "<p><strong>Selected transformation:</strong> $t_txt</p>"; foreach($seqs_array_t as $seq_array_t){ $header_t = $seq_array_t[0]; // Header transformed (_t) $seq_t = $seq_array_t[1]; // Sequence transformed (and also properly HTML tagged, we did that in the previous foreach cycle) echo "<p>$header_t<br>$seq_t</p>"; } echo file_get_contents("html/footer.html"); // Writing the footer HTML to the output page ?> |

You may test the script live here.
Chapter Sections
[pagelist include=”1461″]
[siblings]
WORK IN PROGRESS ON CHAPTER 5!