Keywords: organ transplantation, vaccination, immunology, MHC, MHC-peptide complex, T-Cells, Immunoinformatics, epitope prediction, reverse vaccinology, recombinant vaccines
The Major Histocompatibility Complex (MHC) molecules
Organs transplantation is a procedure that has a long history.
We learn from Wikipedia that:
“The Chinese physician Pien Chi’ao reportedly exchanged hearts between a man of strong spirit but weak will with one of a man of weak spirit but strong will in an attempt to achieve balance in each man.”
Pien Chi’ao died in 310 BC.

We don’t know for sure if Pien Chi’ao’s procedure succeeded as planned. However, we do now know that allotransplants, transplants from one person to another, can easily fail because of a phenomenon known as rejection. In order to succeed, the organ to be transplanted has to be compatible with the receiver. The molecular basis for this compatibility are now well understood, and can be largely tracked down, at the molecular level, to a group of proteins known as “The Major Histocompatibility Complex”, or in short, MHC (link).
MHC molecules, MHC-peptide complexes, T-Cells and the immune response
Let us briefly overview how the MHC molecules work. MHC molecules are are a part of the adaptive immune system of all vertebrates. They are synthesised in the Endoplasmic Reticulum of cells, an intracellular system of membrane vescicles, where they fold, assuming their final tridimensional structure and then travel to the outer cell membrane where they end up exposed on the exterior part of the membrane, so as to be “visible” from the outside of cells.
While MHC molecules are continuously synthesised and make it to the cell outer part, another quite interesting thing happens inside cells: all the cell proteins undergo a turn-over, that is, they are produced, live in the cell for some time, and then undergo degradation. While the turn-over rate can change from one protein (type) to another, such is the fate of all proteins inside cells.
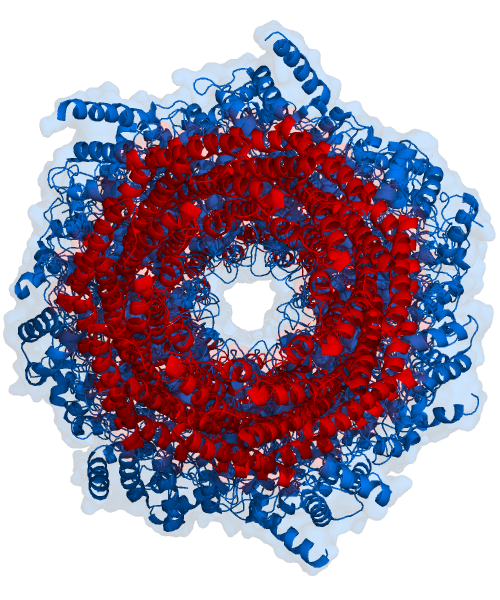
The degradation process is complex, however suffice it to say that there are dedicated molecular complexes inside cells, called “proteasomes”, dedicated to protein degradation by cutting the proteins in pieces, also known as “peptides”.

Think of the proteasome as a tunnel with a pair of scissors in it. And surely enough, by looking at the proteasome structure from the right angle, you can see the hole of the tunnel, as shown in the next figure. Proteins enter the tunnel from one side and while the protein filament travels to the other side cuts are made, so that the protein comes out in pieces (the peptides).
Now the interesting part: the peptides deriving from protein degradation, or at least some of them, will enter the Endoplasmic Reticulum, associate with the MHC molecules by interacting with a special binding groove on the MHC proteins (more on this to come), and finally end up exposed to the outside of the cells, bound to the MHC molecules. This fascinating process literally creates a representation, on the cell surface, of the protein pool contained within the cell, displayed as MHC-peptide complexes. Again, it is important to understand that these MHC-peptide complexes are “fully visible” on the cell surface, from outside the cells.
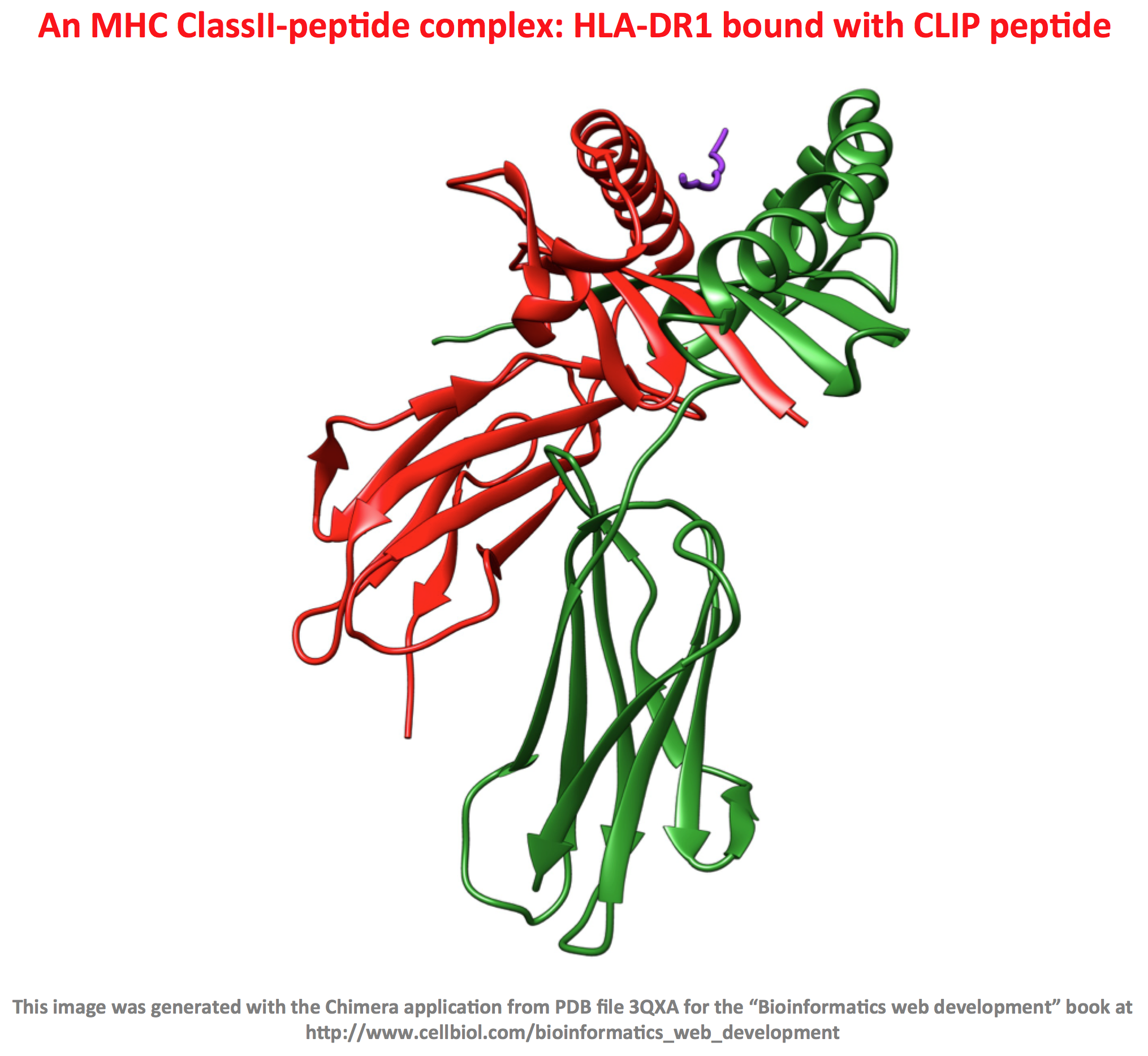
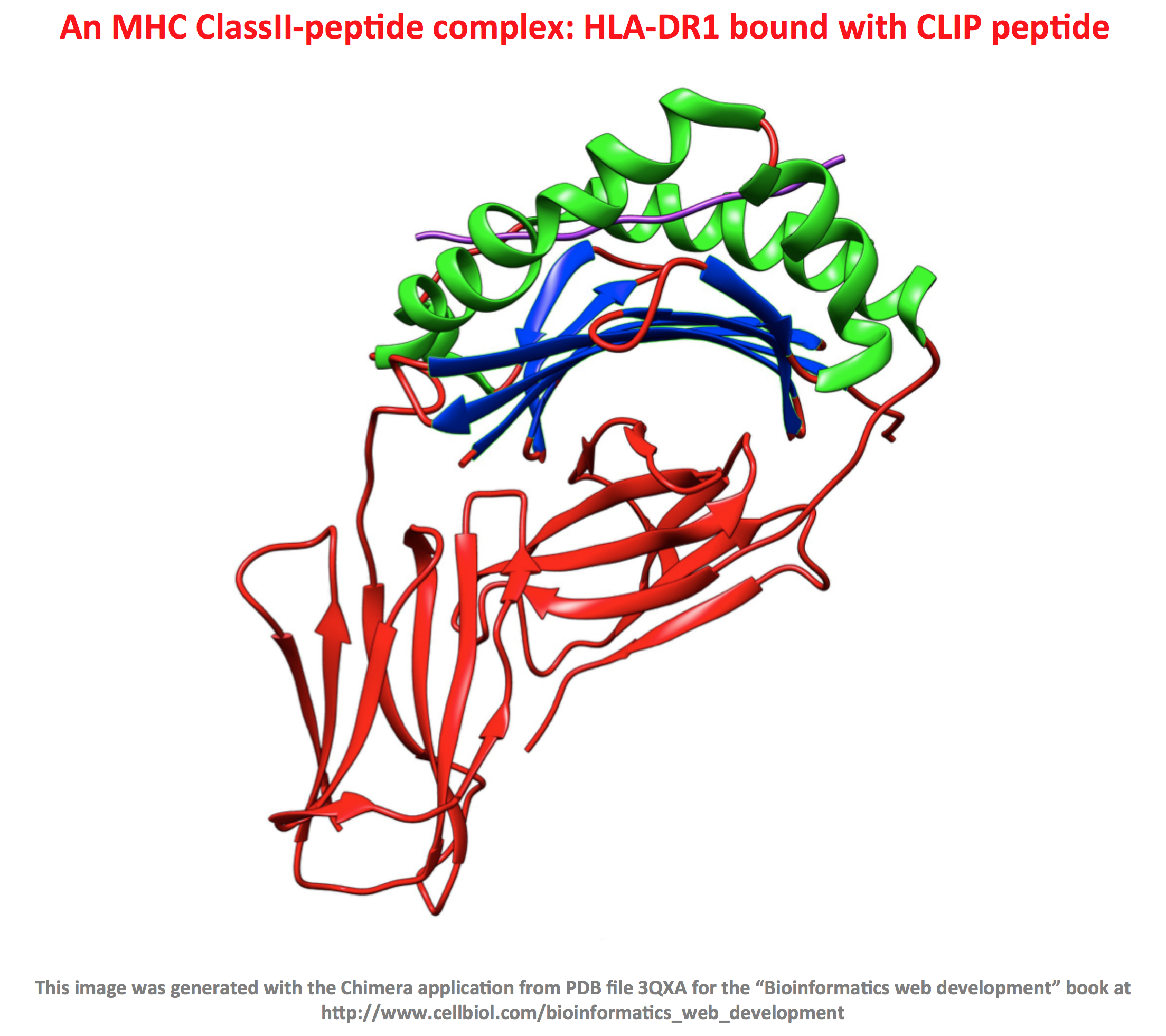
Who’s looking and why are these MHC-peptide molecular complexes so important for transplant compatibility and more in general, the immune response, including defense against infections and cancer?
The above-mentioned process of MHC-peptides complexes exposure on the outer membranes happens virtually in all human cells (except erythrocytes – red blood cells). The “watchers” instead, are a very specialized population of cells belonging to the immune system known as T-Cells. On their surface, T-Cells have a very special receptor, indeed a protein specifically expressed in these cells, called the T-Cell Receptor. In their early life, T-Cell were “trained” to spot, with an extremely high degree of precision, MHC-peptides complexes that legitimately belong to the organism. Indeed the T-Cell Receptor can recognise and bind to these complexes. When T-Cells detect that the MHC-peptide on the surface of a cell in not “legitimate” and looks foreign, they become deadly killers (more specifically, a specialized subset of T-Cells called “cytotoxic T-Cells”, do), releasing a set of proteins called perforins that create a hole in the targeted cell, the one with the suspect MHC-peptide complex on the surface, and then use this hole to inject still another set of proteins called granzymes, that kill the target cell by triggering a process known as apoptosis. The perforins/granzymes mechanism is nicely shown in action in the video included below in this page.
Why are the donor cells detected as foreign by the transplant receiver? As it turns out, the genes that encode MHC molecules are highly polymorphic in the population. Although the number of genes that encode for MHC molecules is limited, there are many variants (alleles) around, and each person has it’s own set of variants.
This makes a lot of sense from the evolutionary point of view, as the variability ensures that when a pathogen, bacteria or virus hits a population, at least some individuals will have an appropriate immune response and survive, ensuring the propagation of the species. In molecular terms, in the population there will be at least some MHC molecules able to bind to the pathogen peptides thereby ensuring that the individuals who carry those MHC variants will elicit a correct T-Cell response to the infection and will survive. So you see how the binding of the peptides to the MCH is central, a key step during the immune response to infections (and cancer).
However there is a downside for transplants, as on transplanting tissues or organs from one individual to another, the chances are high that the MHC molecules of the donor will differ from the ones of the receiver. This will trigger an immune response by the T-Cells of the receiver, that will perceive (correctly) the donor’s tissues as foreign (non-self) and cause a rejection response. So in the case of transplantation, the reason for the reaction of the receiver against the donor’s tissues is related to the difference in the MHC molecules set of the two individuals (rather than to the peptides that bind to the MHC).
What about infections and cancer? If a cell is infected by a virus, the virus proteins, as all the legitimate cellular proteins, will undergo turn over, resulting in peptides that will be displayed on the cell surface thanks to the mechanism we have described above, driven by the MHC molecules. And here you have an MHC-peptide complex that T-Cells do not recognise as “self”. Unusual proteins can be generated in cancer cells, leading to the same result. In these cases, while the MHC molecules are the same in all cells of an individual (this is an approximation, but let’s stand with that for the sake of this overview), it is the associated peptides that change.
Again for infections, or cancer, to be correctly recognised as foreign, a key molecular event is that some foreign or unusual peptide needs to bind to an MHC molecule and be exposed as an MHC-peptide complex on the cell surface. This interaction is nicely showed in the following video that features an animation in which, among other things, the T-Cell Receptor on a T-Cell binds to an MHC-peptide complex on another cell. If the peptide is foreign (red in the video), there will be consequences.
A last bit of valuable information in this overview is the T-Cell receptors are highly variable, each T-Cell has a different one (again an approximation for the sake of this discussion, bear with us). Once a particular T-Cell detects a foreign MHC-peptide complex, for example because the peptide derives from a viral protein, and this particular T-Cell has the right T-Cell receptor to recognise this specific MHC-peptide complex, the T-Cell undergoes replication, also known as “clonal expansion”. This will enhance the capability of the organism to fight this particular infection as it occurs, but also in the future. This expanded clone represents an “immunological memory” of the original exposure event. As a metaphor, we could say that if a soldier proves to be particularly good at spotting or fighting an enemy during a battle, you want more soldiers like him in the army. This clonal expansion constitutes one of the cellular based mechanisms of vaccination. Exposing an individual to a weakened pathogen, or to parts of a pathogen, in other words to a vaccine, can generate a clonal expansion of the T-Cells able to recognise and fight the infection in the future, so that when, maybe years later, the vaccinated individual will come in contact with the pathogen again (maybe the full, live version this time), he will be able to elicit a quicker and more effective immune response and survive or cope better with the infection.
Traditional vaccines, recombinant vaccines and the reverse vaccinology approach

The topic of vaccines is complex and vaccines for different infectious diseases can differ widely in how they are obtained or prepared. We will keep this discussion short and at a very generic level with the only aim to put the T-score application that we will develop later on in an appropriate cultural and scientific context.
In early times, human vaccines were based on non pathogenic versions of the bacteria or viruses that cause an infection. These versions are non pathogenic because they may be heterotypic, that is a variant of the pathogen that naturally does not cause a disease in humans, but is still very similar to the original pathogen, or because they are made by preparations of the actual pathogen organism which has been somehow attenuated or inactivated with a treatment, for example exposure to some chemical agent, so that it is not virulent anymore.
An example of an heterotypic vaccine is cowpox, a virus that normally infects and cause disease in cows. Humans exposed to cowpox become resistant to it’s closely related virus smallpox, which indeed causes smallpox disease in humans. Cowpox is not itself pathogenic to humans, but it is similar enough to smallpox to elicit an immune response that generate an immunological memory in humans able to protect them if they become exposed to smallpox at later times. In short, cowpox is a vaccine for human smallpox, as discovered, among others, by Eduard Jenner who tested the vaccine in 1796 (ref: Wikipedia).

Another example of heterotypic vaccine is the BCG vaccine, produced from Mycobacterium bovis, that protects against human tuberculosis, a result of infection by Mycobacterium tuberculosis.
A classical example of an attenuated vaccine based on the actual human pathogen is the vaccine for rabies developed by Louis Pasteur. The virus was attenuated by growing it in rabbits and then drying the nerve tissue that contained it and proved to be effective in treating a 9 years old to which the vaccine was administered by Pasteur in 1885 (ref: Wikipedia).

The main point we wish to make here is that both heterotypic and attenuated or inactivated vaccines are based on whole viruses or bacteria. While this kind of vaccinations indeed proved to be highly effective and saved so many lives in the human history, the current knowledge of how the immune system works, of what exactly within the whole organisms used for vaccination elicits a protective immune response and the advances in the field of Molecular Biology and Biotechnology could now allow a more rational and “specific” approach to vaccination. The reason being that within an organism, only some parts elicit a protective immune response. A better and “cleaner” vaccine could comprise only those parts rather than the whole organism. Sometimes a single protein from a virus it sufficient to elicit an effective immune response, and thanks to gene cloning and techniques that allow for gene expression and protein purification in bacteria or yeast cells, such a protein (called a “recombinant protein”) can be produced in high amounts, at an high grade of purity, in a way that is virtually entirely independent from the original pathogenic organism.
A successful example of such a vaccine based on the recombinant form of single protein from a pathogenic organism is the Recombivax HB® vaccine against Hepatitis B, made by a preparation of the hepatitis B surface antigen (HBsAg) produced in yeast cells.
Yet, using entire proteins for vaccination may still be an overshot, the reason being that, as it is true that only some proteins from a pathogenic organism elicit a protective immune response, it is also true that within those proteins, only some portions are responsible for such a response, the so-called B-Cell and T-Cell epitopes.
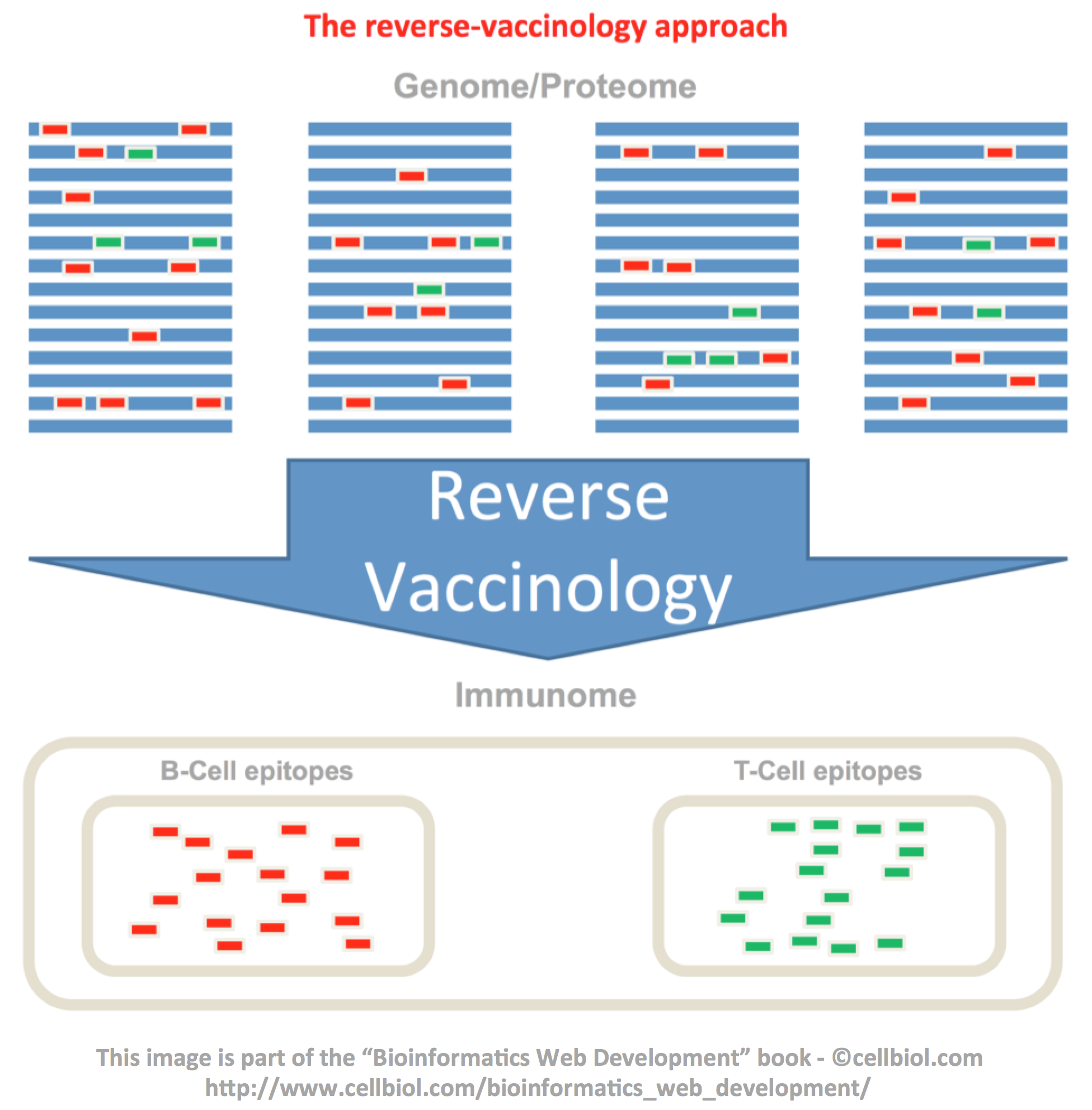
Here comes into play the “Reverse Vaccinology” approach, originally proposed by Rino Rappuoli and used by his research group to identify vaccine candidates against serogroup B Meningococcus (Ref: Science, Curr. Op. Microbiol., Wikipedia).
The rationale of the reverse vaccinology approach is based on these elements:
- It is now very easy and relatively inexpensive to obtain the full genome sequence(s) for pathogenic organisms, bacteria and viruses
- The genome sequence virtually contains the information to compute all the sequences of all proteins of these organisms (the so-called “proteome”)
- The immunoinformatics methods are improving over the years allowing a prediction of which proteins, within the hole proteome, will elicit a protective immune response in the host (including humans)
- Those methods also allow to predict, with increasing degree of precision and reliability, the portions of these proteins able to elicit an antibody (humoral) response, called B-Cell epitopes, and a cellular response, called T-Cell epitopes
- The usage of (some of) these epitopes alone, as opposed to whole pathogens or entire proteins, could in principle elicit a protective immune response and immunological memory, and therefore be used as vaccines
This process is outlined in the following figure.
The following image from Wikipedia outlines the process up to the FDA approval and recommendation phases:
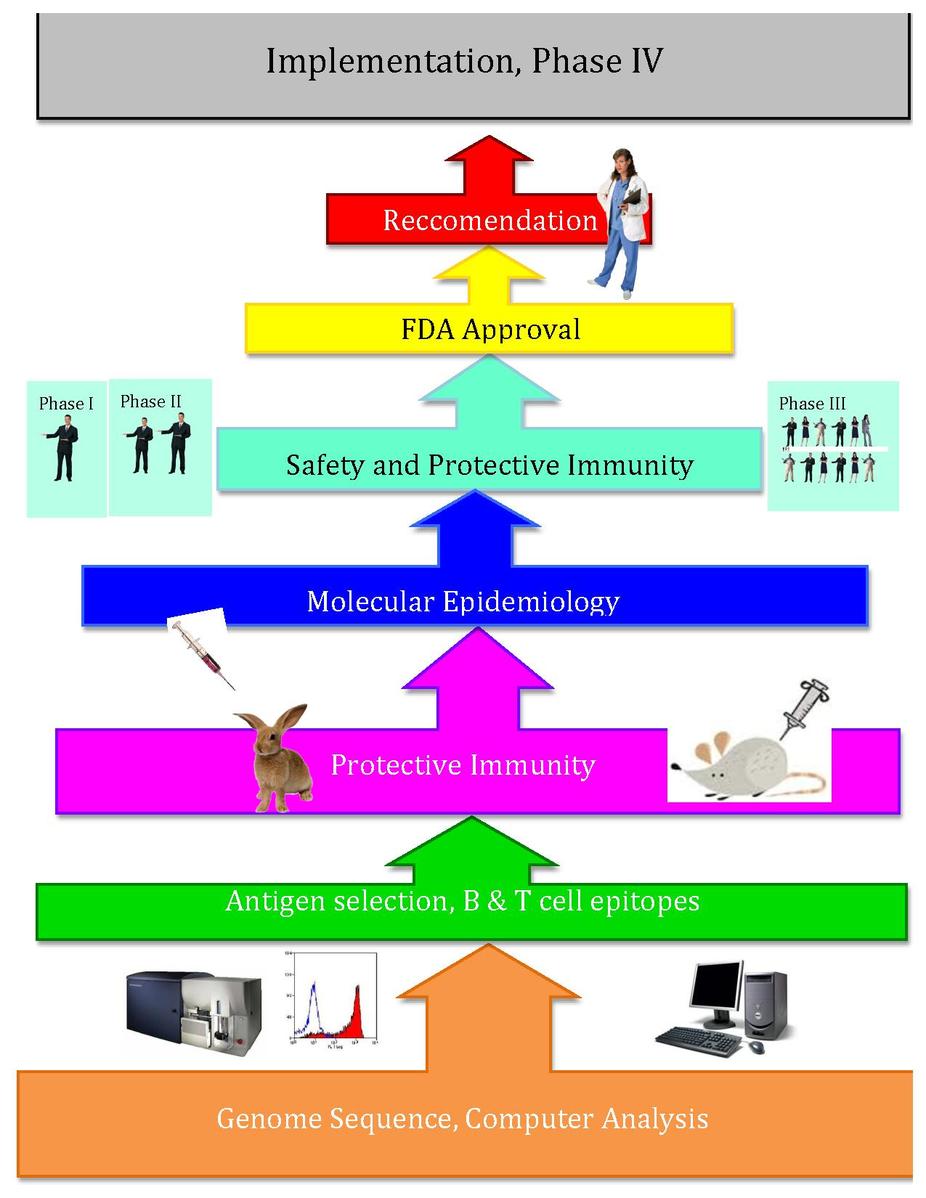
Prediction of T-Cells epitopes
Central to the reverse vaccinology approach is the in-silico prediction of B and T-Cells epitopes from protein sequences. Several research groups are working on the development of such methods with a variety of bioinformatics techniques that take into account different properties of protein sequences and/or structures. For our T-score application we will focus on a method based on scoring matrices published in 2007 in Journal of Biosciences (ref). More precisely, on the part of the method that assigns scores to peptides, 9mers (peptide sequences with a length of 9 amino-acids), based on these scoring matrices.
Scoring matrices for HLA alleles
Thanks to the nice work of Prof. G.P. Raghava and co-workers, the scoring matrices are publicly available at this address. A distinct matrix is available for each of the HLA alleles (MHC protein variants) considered in the original study. Indeed, each MHC variant preferably binds to different peptide sequences. This is why, as already discussed above, it is important that within the human population several variants are available to face a wide range of different viral or bacterial infections and why MHC-peptide binding predictions need a different scoring matrix for each variant. We will limit our T-Score web application to the implementation of three of these matrices, namely the ones for the HLA-A1, A2 and A3 alleles.
As an example, a screenshot of the scoring matrix for the HLA-A1 allele is shown in the figure below.
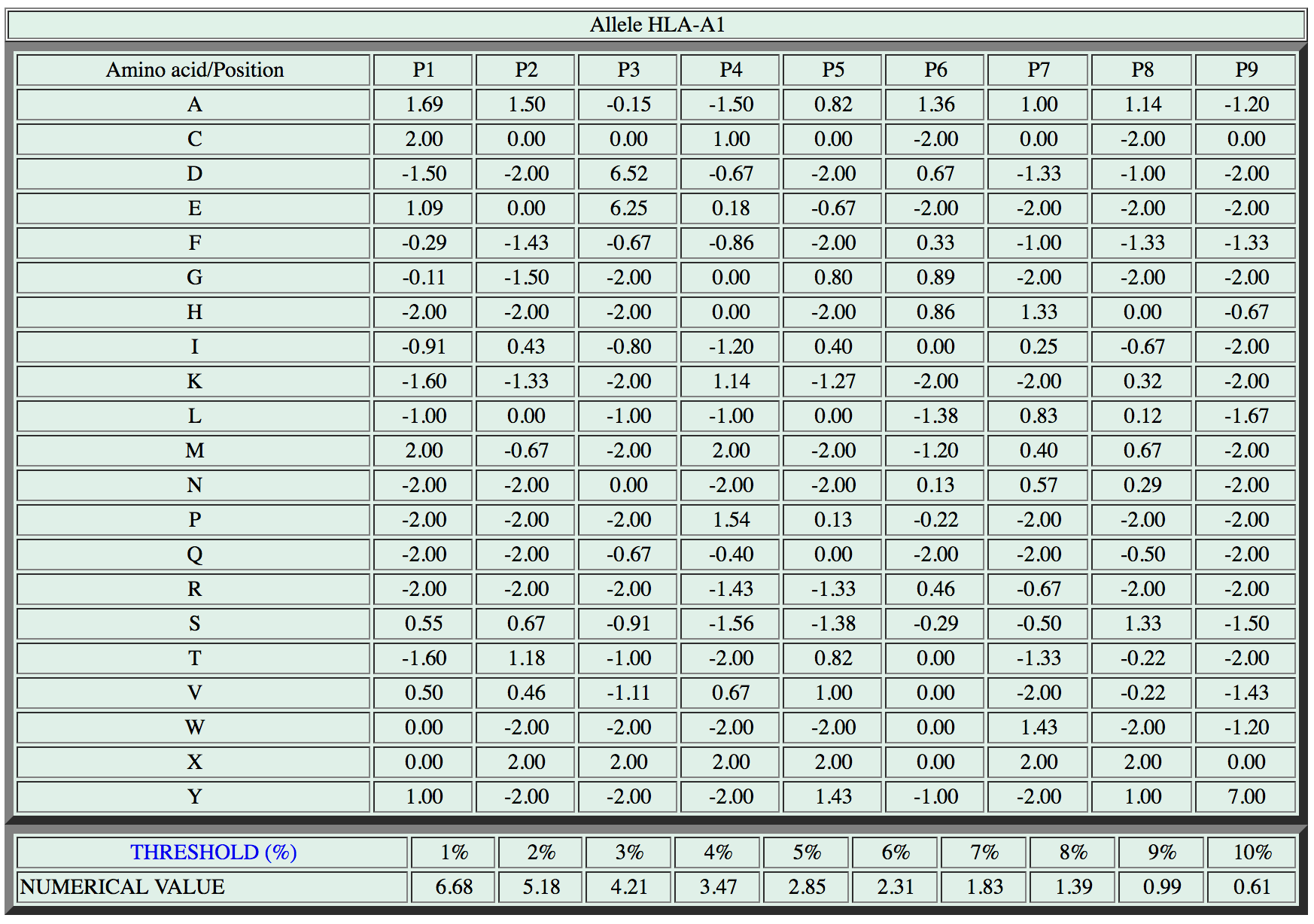
How to use a scoring matrix to assign a score to a peptide
Before implementing any software it is important to understand how to use a matrix to assign a score to a peptide. It is a simple and straightforward task.
Let us consider the following hypothetical 9 amino-acids peptide that we wish to score for HLA-A1 binding (see the figure above for the scoring matrix).
KLDVHRTAC
The matrix is used by looking up the amino-acids scores, one by one, and adding them up. Each amino-acid in a particular position (P1 to P9, positions 1 to 9) is assigned a specific score by the matrix. For our sample peptide:
K in position 1 (P1) is worth -1.6
L in position 2 (P2) is worth 0
D in position 3 (P3) is worth 6.52
V in position 4 (P4) is worth 0.67
H in position 5 (P5) is worth -2
R in position 6 (P6) is worth 0.46
T in position 7 (P7) is worth -1.33
A in position 8 (P8) is worth 1.14
C in position 9 (P9) is worth 0
Let’s add it up:
– 1.6 + 0 + 6.52 + 0.67 – 2 + 0.46 – 1.33 + 1.14 + 0 = 3.86
The peptide score is 6.52. The last two lines of the matrix allow us to rank this peptide among other peptides know to bind the HLA-A1 MHC sequence. The score of our peptide is higher than the 3.47 threshold indicated for the top 4% binders, but lower than the 4.21 threshold indicated for the top 3% binders. So we can rank our sample sequence among the top 4% binders for the HLA-A1 sequence.
In the next section we will write the code for the T-Score web application. With respect to our previous applications examples it will be a somewhat more complex endeavor. Are you ready?
Chapter Sections
[pagelist include=”1461″]
[siblings]
WORK IN PROGRESS ON CHAPTER 5!